Management, analysis and archival of computational data (2002-2006)¶
Computer simulations and computer based data analysis have resulted in a significant increase in scientifit data of all kinds that have to be archived, accessed and processed to be interpreted. While there has been remarkable progress in reducing the storage density of modern hard disks, there is little research into methodologies how to manage that data on the software and userlevel. This project is investigating exactly this issue.
BioSimGrid¶
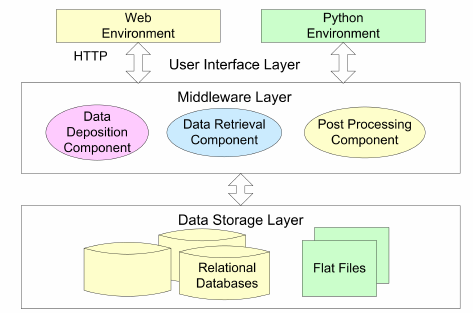
The BioSimGrid project [1] is developing a geographically distributed database for biomolecular simulations to enable users to store and analyse trajectories from a number of different simulation packages. This provides an infrastructure to allow researchers in biochemistry to (i) deposit their simulation results to the system indepent of the simulation software they have used to compute the data, (ii) to retrieve the deposited data at any site of the network and (iii) use the provided analysis tools to carry out post-processing of the deposited data. Some of the advantages of the system are the ability to use the same analysis tools on trajectories from different simulation packages and to carry out cross-trajectory analyses.
Towards the Dynome: Adding a 4th Dimension to the Protein Database by Terascale Simulation¶
The way in which proteins move and flex is critical for their function. Unfortunately, the main experimental method for probing and determining protein structure, X-ray diffraction, provides comparatively little information on how the protein moves. Molecular dynamics computer simulations, on the other hand, are able to yield detailed information on protein dynamics that is very useful, particularly for rational drug design. In this work, large-scale high-performance computers are used to simulate the dynamics of a broad range of the most important protein shapes, or folds. Studies of how specific mutations (for example, drug resistant mutations) affect the protein motions will also be performed. These simulations demonstrate the power and importance of integrating large computing resources. Computer software to control the simulations, store their output, analyse and present the results, and select new simulations to be run, will be demonstrated. These studies are important not only for the computing community itself, but are also particularly timely because of recent development in protein X-ray structure determination. With the arrival of the Diamond Light Source in 2007, and the stated aims of the UK Structural Genomics Consortium (http://www.sgc.ox.ac.uk), the number of new protein structures available will increase significantly.
[1] http://doi.org/10.1039/B411352G
[2] http://doi.org/10.1016/j.future.2005.10.005
[3] http://doi.org/10.1007/978-3-540-69389-5_39